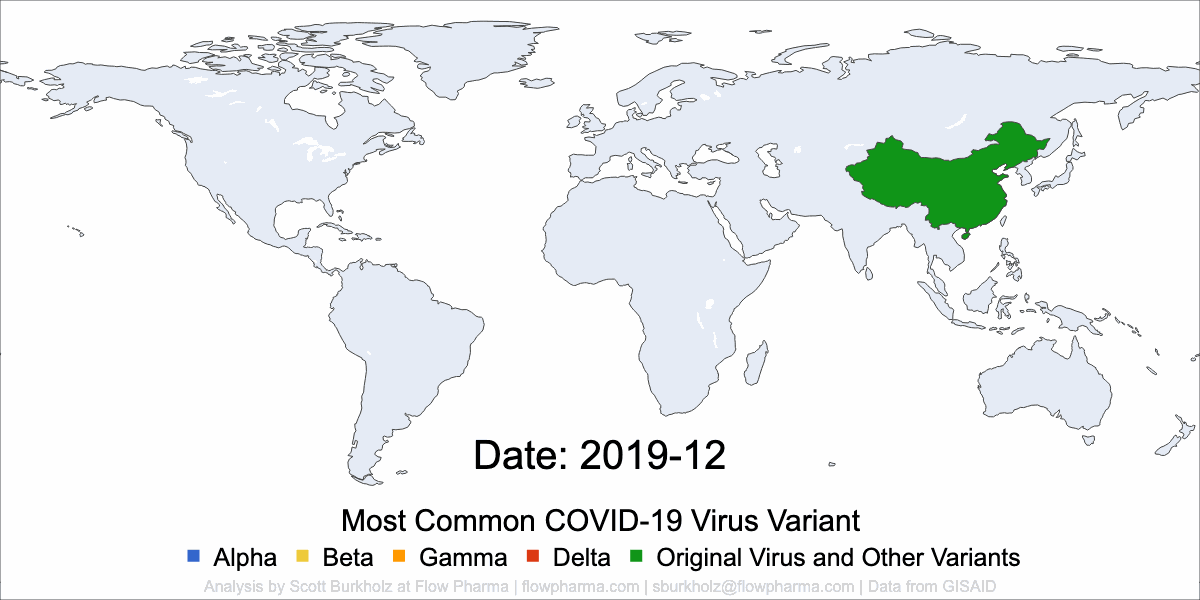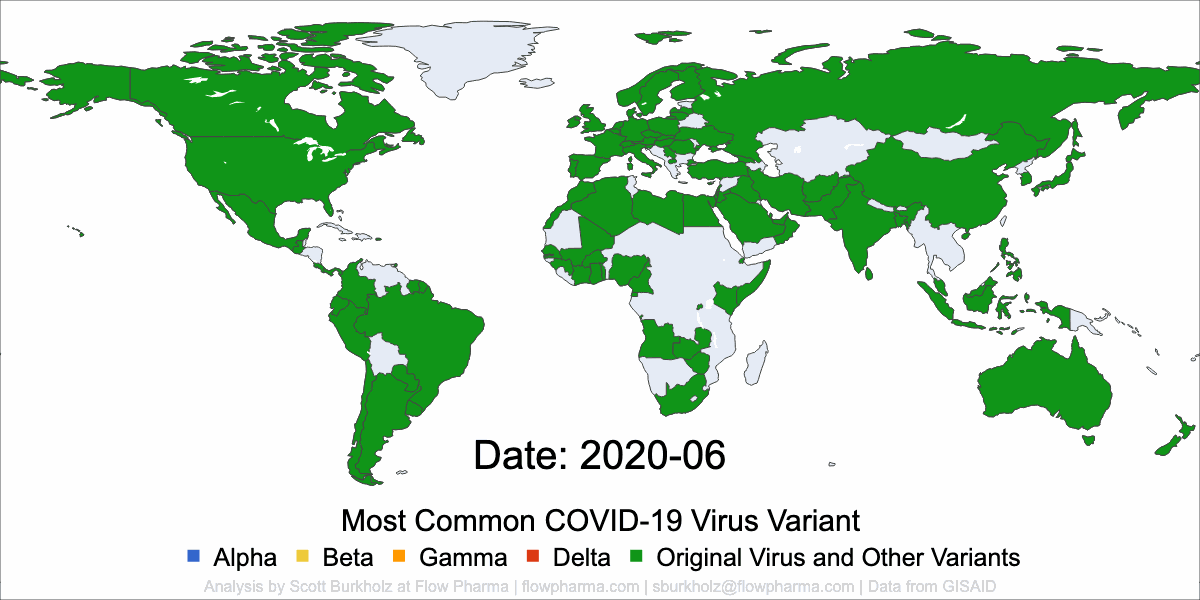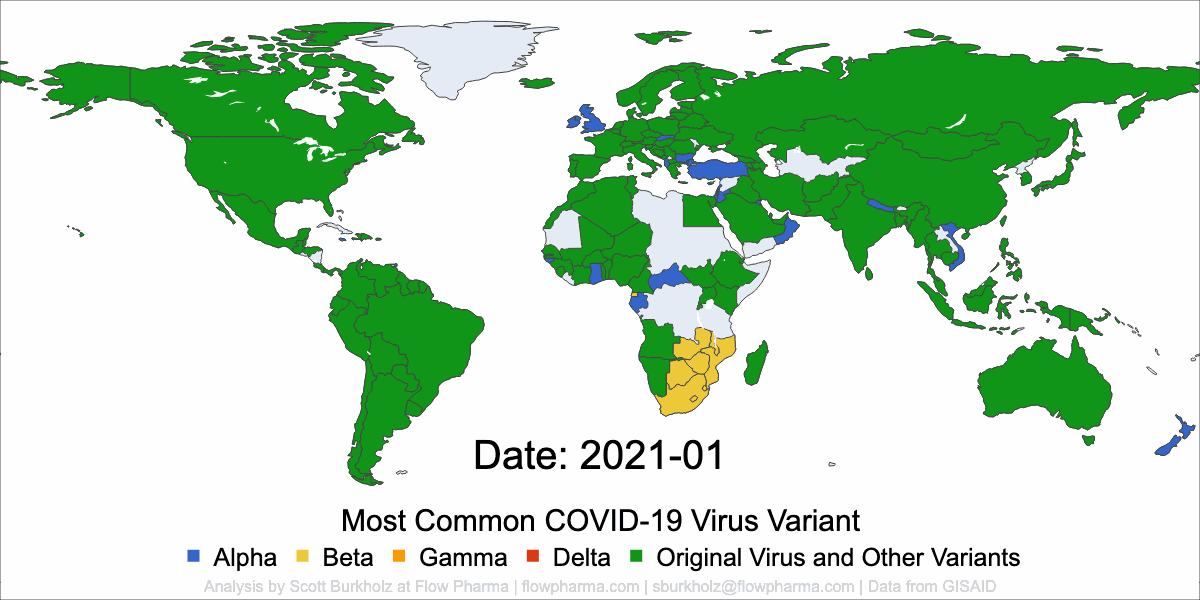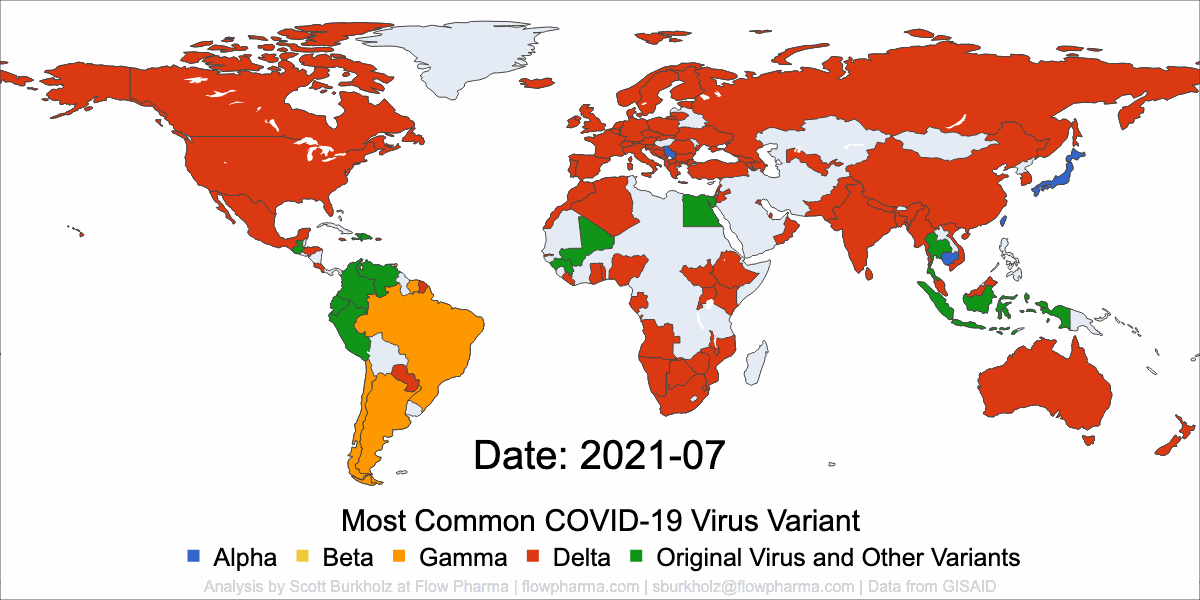
PLEASANT HILL, CA / ACCESSWIRE / May 19, 2021 / Flow Pharma, Inc., a San Francisco Bay Area biotechnology company developing the FlowVax™ peptide vaccine platform technology, today announced the publication of an article describing research performed at the University of Texas Medical Branch at Galveston (UTMB). The article, entitled, “A synthetic peptide CTL vaccine targeting nucleocapsid confers protection from SARS-CoV-2 challenge in rhesus macaques,” was published May,18 2021 in the peer-reviewed journal Vaccines. Dr. Paul Harris and Dr. Trevor Brasel were equally contributing first authors on the paper.
“Several spike antibody evoking vaccines are being deployed in mass vaccination programs. Recent data suggest that as spike mutations increase, these vaccines may lose efficacy” said Paul Harris, Ph.D., Department of Medicine, Columbia University. “We are pleased with the positive results of this COVID-19 study, which were achieved by using a T-cell vaccine that does not target the mutating spike protein.
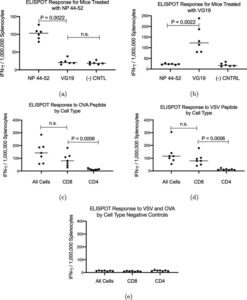
Trevor Brasel, Ph.D., M(ASCP)CM, assistant professor, Department of Microbiology & Immunology, UTMB said, “We are happy to be working with Flow Pharma on the pre-clinical testing phase of vaccines based on the FlowVax platform. Our team has extensive experience working with viruses in pre-clinical models and has been focusing on animal models for COVID-19 since the beginning of the pandemic. This study demonstrated that animals vaccinated with FlowVax COVID-19, and later challenged with SARS-CoV-2, were free of pneumonia-like infiltrates characteristic of COVID-19 infection, and presented with lower viral loads relative to controls.”
Dr. Brasel is author/co-author of over 80 peer-reviewed articles, editorials, book chapters, and presentations on topics including bioaerosol science and vaccine efficacy testing. Dr. Brasel’s ultimate goal is the development of high-profile FDA-compliant medical countermeasures specific to biodefense-related and emerging infectious diseases.
“We designed FlowVax COVID-19 to fight SARS-CoV-2 infections with killer T-cells rather than antibodies,” said Charles Herst, Ph.D., chief science officer, Flow Pharma. “This allows us to target the portions of the virus least likely to mutate, mitigating the risk of viral escape from vaccines that lead to a primarily antibody-driven response. In this study, we showed that a room-temperature-stable peptide vaccine targeting the SARS-CoV-2 nucleocapsid protein can provide protection against COVID-19 infection in nonhuman primates.”
“We are currently manufacturing clinical trial test doses of FLOVID-20, a version of FlowVax COVID-19 optimized for mass production,” stated Reid Rubsamen, M.D., M.S., CEO Flow Pharma, Inc. “The next step will be to conduct clinical trials of FLOVID-20 as a therapeutic for patients with acute and chronic COVID infections.”
About Flow Pharma
Flow Pharma, Inc. engineers and manufactures next generation T-cell stimulating biomedicines to treat and prevent viral diseases and cancers. Current products under development include FLOVID-20, to treat and prevent COVID-19, Ebola and Marburg virus vaccines and cancer vaccines to treat patients with breast cancer and cancers caused by human papilloma virus, including cervical cancer and throat cancer. Flow Pharma uses a proprietary mix of artificial intelligence, bioinformatics, and synthetic biochemistry to design and manufacture its expanding product portfolio.
About the University of Texas Medical Branch and the Galveston National Laboratory
The Galveston National Laboratory (GNL) is a sophisticated high containment research facility that serves as a critically important resource in the global fight against infectious diseases. The GNL is located on the campus of the University of Texas Medical Branch and operates under the umbrella of UTMB’s Institute for Human Infections and Immunity. The GNL is an anchor lab of the NIAID Biodefense Laboratory Network and is one of only two National Laboratories with Biosafety Level 4 (BSL-4) capabilities located on a U.S. university campus.
Forward Looking Statements
This press release may contain forward-looking statements, including information about management’s view of Flow Pharma, Inc. (“the Company”), future expectations, plans and prospects. In particular, when used in the preceding discussion, the words “believes,” “expects,” “intends,” “plans,” “anticipates,” or “may,” and similar conditional expressions are intended to identify forward-looking statements. Any statements made in this presentation other than those of historical fact, about an action, event or development, are forward-looking statements. These statements involve known and unknown risks, uncertainties and other factors, which may cause the results of the Company, its subsidiaries and concepts to be materially different than those expressed or implied in such statements. Unknown or unpredictable factors also could have material adverse effects on the Company’s future results.
The forward-looking statements included in this presentation are made only as of the date hereof. The Company cannot guarantee future results, levels of activity, performance or achievements. Accordingly, you should not place undue reliance on these forward-looking statements. Finally, the Company undertakes no obligation to update these statements after the date of this release, except as required by law, and also takes no obligation to update or correct information prepared by third parties that are not paid for by Flow Pharma, Inc.
Contact:
Investor Relations
Flow Pharma, Inc.
4829 Galaxy Parkway
Suite K
Warrensville Heights, OH 44128
925-708-8000
www.flowpharma.com
[email protected]
Published on Accesswire