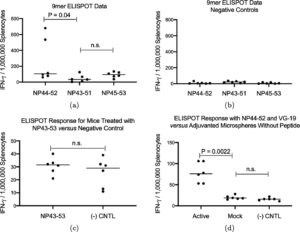
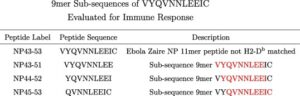
After successfully completing STTR Phase I funded design of a Marburg virus prophylactic countermeasure vaccine, Flow Pharma has received a follow-on Phase II STTR contract award to transition into vaccine manufacturing and pre-clinical testing.
Flow Pharma today announced that the Company has received a Phase II STTR contract award to continue developing a protective vaccine against Marburg virus. This award follows Flow Pharma successfully completing the Phase I STTR contract from the United States Department of Defense for the same project. Flow Pharma is working in collaboration with Dr. Trevor Brasel and his team at the University of Texas Medical Branch (UTMB) in Galveston, Texas.

“We are happy to be working with Flow Pharma on the pre-clinical testing phase of their FlowVax Marburg development program. Our team has extensive experience working with Marburg virus and similar viruses like Ebola in various pre-clinical models. We are very fortunate at UTMB in that we have access to unique BSL-4 laboratories, including the Galveston National Laboratory and the Robert E. Shope, MD, Laboratory, that allow us to conduct studies like these safely and at a high level of quality to ensure data integrity for sponsors like Flow Pharma,” said Trevor Brasel, Ph.D., M(ASCP)CM, Assistant Professor, Department of Microbiology and Immunology, UTMB.
Dr. Brasel is author/co-author of over 70 peer-reviewed articles, editorials, book chapters, and poster/podium presentations on a broad range of topics including bioaerosol science and vaccine efficacy testing against select agents. To date, no vaccines or therapeutics have been approved for human use against Marburg virus. Dr. Brasel’s ultimate goal is FDA approval of high-profile medical countermeasures specific for biodefense-related and emerging infectious diseases.
“We completed a design for FlowVax Marburg using computer modeling and in-vitro testing during the first phase of this STTR funded program. We look forward to the second phase of the contract awarded today which will take us into manufacturing and testing FlowVax Marburg vaccine in a pre-clinical model,” said C.V. Herst, Ph.D., Flow Pharma Chief Science Officer.
“We appreciate the support from the United States Department of Defense for this important work. Marburg virus has been weaponized by foreign actors and represents both a military and civilian threat. We are pleased to have successfully completed our STTR Phase I contract, leading to the follow-on contract award we are announcing today,” said Flow Pharma CEO, Reid Rubsamen, M.D.
Notice
This material is based upon work supported by the US Army Contracting Command – Aberdeen Proving Ground – Natick Contracting Division under Contract No. W911QY-20-C-0057.
Any opinions, findings and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the US Army Contracting Command – Aberdeen Proving Ground – Natick Contracting Division.
About the University of Texas Medical Branch and the Galveston National Laboratory
The Galveston National Laboratory (GNL) is a sophisticated high containment research facility that serves as a critically important resource in the global fight against infectious diseases. The GNL is located on the campus of the University of Texas Medical Branch and operates under the umbrella of UTMB’s Institute for Human Infections and Immunity. The GNL is an anchor lab of the NIAID Biodefense Laboratory Network and is one of only two National Laboratories with Biosafety Level 4 (BSL-4) capabilities located on a U.S. university campus.
About Flow Pharma
Flow Pharma, Inc. is a San Francisco Bay Area based, biotechnology company using artificial intelligence to guide the selection of neoantigen peptide targets on cancer cells or virus-infected cells for attack by the patient’s own, native immune system. These peptides can then be loaded into the FlowVax platform for administration by injection or inhalation.
Flow Pharma, Inc. is currently engaged in pre-clinical testing of FlowVax COVID-19, a vaccine for protection against SARS-CoV-2 at the University of Texas Medical Branch (UTMB) in Galveston, Texas and is also preparing to test therapeutic cancer vaccines targeting neoantigens found in various cancers. Neoantigens are small peptide markers expressed on cancer cells as a result of a cancer-causing viral infection or mutation of the DNA in normal cells that subsequently contribute to transformation of the normal cells into cancer cells.
Forward Looking Statements
This press release may contain forward-looking statements, including information about management’s view of Flow Pharma, Inc. (“the Company”), future expectations, plans and prospects. In particular, when used in the preceding discussion, the words “believes,” “expects,” “intends,” “plans,” “anticipates,” or “may,” and similar conditional expressions are intended to identify forward-looking statements. Any statements made in this presentation other than those of historical fact, about an action, event or development, are forward-looking statements. These statements involve known and unknown risks, uncertainties and other factors, which may cause the results of the Company, its subsidiaries and concepts to be materially different than those expressed or implied in such statements. Unknown or unpredictable factors also could have material adverse effects on the Company’s future results. The forward-looking statements included in this presentation are made only as of the date hereof. The Company cannot guarantee future results, levels of activity, performance or achievements. Accordingly, you should not place undue reliance on these forward-looking statements. Finally, the Company undertakes no obligation to update these statements after the date of this release, except as required by law, and also takes no obligation to update or correct information prepared by third parties that are not paid for by Flow Pharma, Inc.